
If you or someone you love was diagnosed with HbSC sickle cell disease, there is a reasonable chance you were told by a clinician, a pamphlet, or a well-meaning relative that you had the milder form of sickle cell disease. Something to manage, yes.
Something to monitor. But not quite as serious as the HbSS patients in the waiting room. That framing is clinically inaccurate, and it has real consequences for how people with HbSC disease are screened, treated, and counselled globally.
Key Takeaways
- HbSC is the second most common form of sickle cell disease globally, accounting for roughly 30% of all SCD cases.
- Eye disease (retinopathy) develops in 43% of HbSC patients versus 14% of HbSS patients over a 20-year period, according to a Jamaican cohort study.
- HbSC causes its damage through a different mechanism than HbSS: thicker, denser blood rather than faster red cell destruction.
- There was no proper animal research model for HbSC disease until 2025, meaning 75 years of research largely excluded it.
- AS x AC couples have a 25% chance of having an HbSC child with every pregnancy.
UNDERSTANDING YOUR GENOTYPE
What Is HbSC Disease and How Is It Different from HbSS?
HbSC happens when a person inherits one sickle gene (HbS) from one parent and one haemoglobin C gene (HbC) from the other.
In HbSS disease, both genes are the sickle type. In HbSC, only one is.
Here is where it gets important: having one sickle gene alongside HbC is not the same as having sickle cell trait (AS), which causes no disease.
The HbC gene causes red blood cells to lose water and shrink. Smaller, denser cells concentrate the sickle haemoglobin in a way that still causes it to clump and block blood flow, just through a different route than HbSS.
UNIQUE INSIGHT: HbSS disease destroys red blood cells quickly, causing severe anaemia. HbSC patients tend to have higher blood counts, and that is often read as the body "coping well."
What it actually reflects is thicker, stickier blood that flows poorly through tiny vessels, particularly in the eyes, joints, and spleen.
A reassuringly normal blood count can mask serious disease activity.

What Complications Does HbSC Cause?
Eye Disease: More Common in HbSC Than HbSS
In a 20-year prospective study of Jamaican patients, proliferative sickle cell retinopathy developed in 43% of HbSC patients compared to 14% of HbSS patients.
This is a condition where abnormal blood vessels grow inside the eye and can lead to bleeding and vision loss if not caught and treated.
The American Academy of Ophthalmology confirms that serious eye disease affects up to 40% of HbSC patients and 20% of HbSS patients.
The thicker, slower-moving blood of HbSC disease creates a sustained environment that stimulates abnormal vessel growth in the retina. Annual eye screening is not optional for someone with HbSC.
Joint and Bone Damage
HbSC disease is associated with avascular necrosis, a condition where the blood supply to a bone joint is cut off and the bone tissue begins to die.
This most commonly affects the hips and shoulders.
Recent cohort studies, including the Sickle Cell Disease Implementation Consortium registry, have shown that HbSC patients experience higher rates of avascular necrosis than previously appreciated.
Spleen Complications
Unlike HbSS, where the spleen tends to destroy itself over time, HbSC patients often retain their spleen into adulthood.
This means they remain at risk for acute splenic sequestration, where large amounts of blood suddenly pool in the spleen.
This is a medical emergency and can occur in HbSC patients at older ages than most clinicians expect.
Who Is at Risk of Having an HbSC Child?
HbSC disease can only arise when one parent carries the sickle cell trait (HbAS) and the other carries haemoglobin C trait (HbAC).

With every pregnancy, such couples face a 25% chance of having a child with HbSC disease.
Importantly, HbAC carriers have no symptoms and are frequently unaware they carry the gene at all.
HbAC is most common in West African populations, particularly in Ghana, Burkina Faso, Ivory Coast, and Nigeria, as well as in Afro-Caribbean and some North African communities.
Sickle cell testing before marriage is essential. The Eloheh Rapid Test Kit provides accessible, reliable genotype screening. Get tested today.
What Should HbSC Patients Be Asking?
A diagnosis of HbSC disease should open a conversation, not close one.
Here are questions worth raising with your doctor:
- Has a baseline eye exam been done, and is annual eye screening in place?
- Have the hips and shoulders been assessed for early bone damage?
- Is the spleen being monitored, and does the family know the signs of an acute splenic event?
- Has a full hormone and growth assessment been done for any adolescent patients?
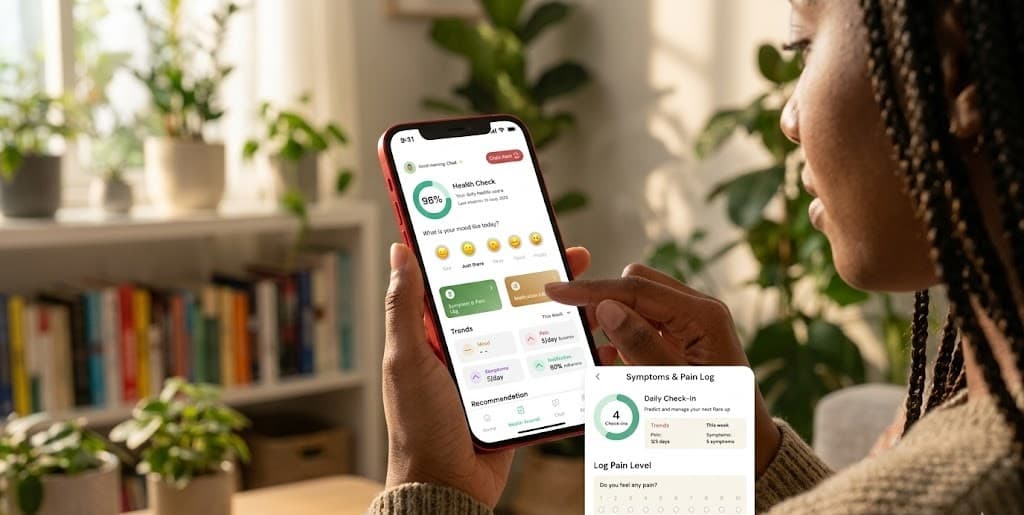
Tracking day-to-day symptoms matters too. Pain patterns, energy levels, and changes in how you feel between appointments all provide important clinical data.
The Eloheh app was built for exactly this: log and track your symptoms at elohehkits.com.
For natural, evidence-based support for people living with sickle cell disease, the Healing Blends Global range was developed specifically around the physiological demands of SCD. EvenFlo has shown 93% effectiveness in a clinical study.
Frequently Asked Questions
Is HbSC really a serious form of sickle cell disease?
Yes. HbSC causes significant health problems, with higher rates of eye disease, spleen complications, and joint damage than was previously recognised.
The label of "mild" originated from comparing HbSC to HbSS, not from comparing it to healthy individuals. By any reasonable standard, HbSC is a serious, lifelong condition requiring active monitoring.
Why do people with HbSC get more eye disease than HbSS?
HbSC produces thicker, denser blood that flows slowly through the tiny blood vessels in the retina.
This creates a low-oxygen environment that stimulates abnormal vessel growth. In HbSS, blood vessels tend to become fully blocked and shut down, which paradoxically prevents the same kind of growth.
This is sometimes called the "sickle cell paradox": the disease that appears less severe systemically carries a significantly higher risk of serious eye complications.
How do I know if I carry the HbC gene?
You won't know without testing. HbAC carriers have no symptoms at all. If you are of West African descent, particularly Ghanaian, Nigerian, or from Burkina Faso, there is a meaningful chance you could carry it. Start with the Eloheh Rapid Test Kit.
Can HbSC patients use natural supplements to support their health?
Yes. Targeted nutritional support, particularly for circulation and inflammation, is relevant for HbSC as it is for other forms of SCD.
Was HbSC included in sickle cell research?
The first proper animal research model for HbSC disease was only published in 2025, more than 75 years after the condition was first described.
This research gap means decades of drug development were conducted without a model that represented HbSC disease at all.
The evidence base for this specific genotype is still catching up.
This article is for educational purposes only and does not constitute medical advice. Always consult a qualified haematologist or healthcare provider for personal medical decisions.
elohehkits.com | @elohehkits